
Enfermedades hereditarias y congénitas
renales felinas
-Introducción
Las enfermedades congénitas son aquellas enfermedades estructurales o funcionales que están presentes en el momento del nacimiento, aunque en muchos casos no se manifiesten hasta los primeros años de vida del animal. La etiología varía y pueden estar provocadas por diversos factores externos que afecten durante el desarrollo embrionario o fetal, o pueden ser el resultado de factores genéticos aleatorios o heredados. Algunas enfermedades congénitas pueden ser hereditarias. Las enfermedades hereditarias son aquellas que se transmiten de los progenitores a su descendencia siguiendo distintos patrones de herencia, debido a mutaciones en la secuencia del ADN, que determinan modificaciones proteicas con la consiguiente alteración de su funcionalidad biológica.
Son pocas las enfermedades de las que conocemos con certeza su patrón de herencia y las mutaciones concretas que lo provocan. Las enfermedades renales congénitas son poco frecuentes, exceptuando la poliquistosis renal (PKD) para la que afortunadamente se dispone de pruebas diagnósticas fiables para su identificación temprana.
■ Características clínicas
La frecuencia real de estas nefropatías y su espectro clínico completo, todavía no se han caracterizado adecuadamente, pero en la Tabla 1 se muestran las nefropatías familiares y hereditarias reconocidas. A efectos prácticos, el síndrome clínico bajo el cual se engloban la mayoría de ellas, es la enfermedad renal crónica (ERC) con las alteraciones hematológicas, bioquímicas, urinarias y clínicas, propias de la disfunción renal.
Los signos clínicos y la edad de presentación varían dependiendo de la severidad del proceso. La aparición de los signos suele ser insidiosa y progresiva y a veces no son advertidos claramente por el propietario hasta que la enfermedad es casi terminal. En algunas enfermedades la muerte sobreviene a los pocos meses de vida después de padecer signos como anorexia, poliuria/polidipsia (PU/PD), retraso en el crecimiento, osteodistrofia renal, anemia, letargia y diversos signos gastrointestinales. En otras como en la poliquistosis o la amiloidosis, ya sea por la penetración parcial o por la naturaleza de la enfermedad en sí, la función renal inicialmente es normal y los gatos pueden vivir durante años, progresando hacia la enfermedad renal con la aparición de signos como PU/PD.
El desarrollo de fallo renal a una temprana edad suele indicar una etiología congénita, aunque los animales jóvenes pueden verse también afectados por procesos adquiridos, de forma que en tan solo 2 meses sus riñones pueden sufrir cambios estructurales hacia un riñón terminal.
Por otra parte, en muchas de estas enfermedades hereditarias, los riñones pueden ser normales al nacimiento y la enfermedad va progresando según avanza la edad, no evidenciándose los signos hasta edades tardías.
El diagnóstico presuntivo se puede establecer basándose en un procedimiento diagnóstico adecuado que incluya la anamnesis, pruebas laboratoriales y técnicas de diagnóstico por imagen (Tabla 2). El diagnóstico definitivo suele requerir de la demostración de las lesiones características en las muestras de riñón obtenidas mediante biopsia o en la necropsia.
Disponer de la biopsia renal sería de gran interés en todos los pacientes con fallo renal, pero esto puede no estar justificado en pacientes en estadios avanzados, ya que el paciente necesita todo el parénquima renal funcional restante y la evaluación clínica suele bastar para establecer un diagnóstico presuntivo y su tratamiento médico adecuado.
Por otra parte, son muchos los pacientes que se diagnostican ya en fases muy avanzadas, por lo que las lesiones primarias no se pueden identificar, y lo que predomina son la fibrosis, los cambios degenerativos e inflamatorios secundarios, característicos de estadios terminales.
Las técnicas de diagnóstico molecular han supuesto un gran avance en el estudio de estas enfermedades, sin embargo, no es posible en todos los casos realizar pruebas genéticas específicas, ya que hay entidades en las que aún no se conoce el gen responsable. Las ventajas del diagnóstico molecular son innumerables, pues además de la confirmación diagnóstica, permite el diagnóstico presintomático a edades tempranas, lo que posibilita descartar al individuo afectado de los planes reproductivos.
No existe un tratamiento eficaz para las nefropatías congénitas. Se benefician de las mismas estrategias de manejo de los pacientes con ERC (disminución de signos urémicos y medidas nefroprotectoras), pero con la dificultad añadida de que en animales muy jóvenes las dietas indicadas para ERC no cubren todas las necesidades nutritivas de crecimiento. En su lugar se sugiere que podría ser de mayor utilidad y más adecuado la administración de quelantes de fósforo.
-Enfermedad Poliquistica Renal (PKD)
La poliquistosis renal felina (PKD), también denominada poliquistosis renal autosómica dominante, es la enfermedad renal hereditaria más frecuente en la especie felina. Se observa fundamentalmente en gatos de raza Persa, en todas aquellas razas que han incluido en sus planes de cría una línea Persa para la adquisición de un determinado rasgo y, de forma esporádica, en otras razas.
En otras razas, tales como el American Shorthair, Siamés, American Curl y Scottish Fold se describen prevalencias de hasta un 16%.
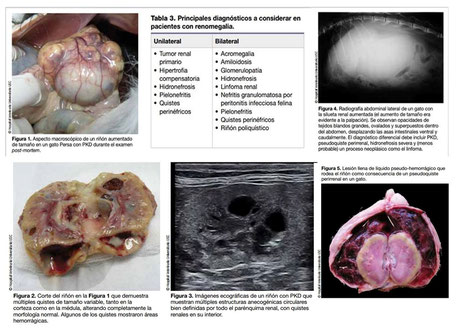
La PKD es una enfermedad monogénica que se caracteriza por la presencia de múltiples quistes renales (Figura 1) más o menos desarrollados y que provocan destrucción del parénquima renal, aunque la progresión varía individualmente.
La PKD se hereda de forma autosómica dominante con penetración completa, es decir, solo una mutación del gen es necesaria para causar la enfermedad y hasta la fecha nunca se ha identificado un gato con dos alelos, pues la combinación de homocigoto se considera letal.
En los planes de cría esto se traduce en el hecho de que si un progenitor se encuentra afectado, la descendencia tiene un 50% de probabilidades de heredar el gen mutado.
Si los dos progenitores poseen una copia del gen mutado las probabilidades se incrementan hasta un 66%, teniendo en cuenta que los embriones con dos copias morirían antes de nacer. En todos los gatos Persas afectados se ha identificado la misma mutación. La mutación es simple; una citosina localizada en el exón 29 es sustituida por una adenina (mutación de transversión). Este simple cambio de bases genera una producción insuficiente de policistina para poder asegurar una función renal normal.
La policistina es una glicoproteína de membrana ubicada en las células del epitelio ciliar que recubre los túbulos renales, y controla la proliferación celular y el mantenimiento de las células tubulares en un estado de diferenciación terminal. La disminución de la policistina por debajo de un nivel crítico da como resultado un cambio fenotípico que se caracteriza por la incapacidad para mantener la polaridad celular, aumento de la tasa de proliferación y apoptosis, la expresión de un fenotipo secretorio y la remodelación de la matriz extracelular, provocando así la aparición de quistes.
El desarrollo de los quistes renales comienza desde la etapa embrionaria y continua durante toda la vida del individuo. Es posible que al igual que ocurre en personas, existan fases diferenciadas de la quistogénesis; una fase sería la de iniciación dependiente de la mutación genética y la otra, una fase de crecimiento independiente de la mutación.
A nivel histológico los quistes se originan como dilataciones focales de los túbulos renales y luego pierden la conexión con estos. En los estadios iniciales el parénquima renal presenta una apariencia relativamente normal, pero en los estadios terminales los riñones adquieren un gran tamaño, presentan innumerables quistes llenos de líquido y contienen zonas parcheadas aisladas de parénquima relativamente normal rodeado de abundante tejido fibroso. En las personas existe una forma autosómica recesiva de PKD que provoca una enfermedad grave de los riñones y de las vías biliares. Se identificó una enfermedad similar en 6 gatitos de 4 camadas familiarmente emparentadas, que mostraron distensión abdominal evidente y murieron antes de las 7 semanas de edad. La necropsia de estos animales mostró PKD y quistes en los conductos biliares.
Los gatos afectados de PKD pueden no mostrar signos clínicos relacionados con la enfermedad o presentar varios signos diferentes. Los quistes renales son responsables de muchas complicaciones tales como hematuria, infecciones de las vías urinarias, cuadros sépticos por infección secundaria de los quistes, pero la complicación más grave es el fallo renal por la destrucción gradual del parénquima, por la expansión de los quistes (Figura 2). La enfermedad renal puede ocurrir a cualquier edad pero es más frecuente entre los 3-7 años de edad.
■ Diagnóstico
Se diferencian en este sentido dos tipos de pacientes:
Aquellos sometidos a estudios para la detección temprana de la enfermedad y aquellos en los que se detecta la enfermedad por sus consecuencias.
Cuando el diagnóstico se hace de forma precoz, la ecografía y los test genéticos son las herramientas fundamentales. Los estudios indican que una vez que el gato tiene 10 meses de edad, la sensibilidad de la ecografía para la detección de la PKD es tan alta como el 95%. Los falsos negativos pueden deberse a presencia de quistes pequeños, inexperiencia del operador o quizá por la presencia de quistes en una localización medular cuya ecogenicidad es similar a la del quiste. Los animales se consideran positivos para PKD cuando se identifica en al menos uno de los riñones una estructura anecogénica mayor de 2 mm de diámetro.
En la actualidad, el avance de los equipos en las técnicas ecográficas ha supuesto un aumento en la sensibilidad diagnóstica incluso a edades tan tempranas como 6-8 semanas de vida, no obstante su ausencia no descarta que la enfermedad se desarrolle a edades más avanzadas.
Pese a la sensibilidad de la ecografía y su utilidad para valorar la progresión de los individuos afectados, para establecer un diagnóstico precoz en individuos potencialmente reproductores, el estudio genético tiene mayores ventajas porque los pacientes pueden evaluarse fácilmente a edades tempranas con un hisopo bucal o con una muestra de sangre. El gen responsable de la enfermedad poliquística se describió en gatos el año 2005 y para su identificación se usan técnicas de amplificación de la cadena de polimerasa (PCR). Al ser una mutación idéntica por descendencia se puede emplear en todos los individuos afectados. No obstante, los test genéticos pueden no identificar todas las formas de enfermedad poliquística en gatos. En estudios recientes, un pequeño porcentaje de gatos con alteraciones ecográficas e histológicas compatibles con PKD poseían el genotipo original silvestre.
Con independencia de que el animal no presente signos, si se ha identificado la PKD, se debe comenzar una monitorización anual o bianual según el grado de afectación de cada individuo; la mejor forma de valorar la progresión de la enfermedad es la ecografía.
En personas, el aumento del tamaño renal y de los quistes es el factor predictivo más importante de pérdida de la función renal. No obstante existen pacientes donde la resistencia vascular parece jugar también un papel destacado y explica aquellos casos en los que el fallo funcional es desproporcionado respecto a la gravedad de la enfermedad quística.
En gatos, si los quistes progresan rápidamente podrían acelerar el desarrollo de los signos clínicos. Posiblemente la gravedad del proceso se debe tanto al desarrollo de un número mayor de quistes a una edad temprana como a una mayor velocidad de crecimiento de los quistes ya existentes. Se estima, que cuando el tejido ocupado por los quistes supera el 75%, la PKD pueda ser la causa potencial de ERC, aunque debe tenerse en cuenta que en animales de edad avanzada puede que existan otros factores que ejerzan un papel sumatorio en la merma dela funcionalidad.
En pacientes enfermos, además de las alteraciones propias que caracterizan la ERC en sus diferentes estadios (según sistema IRIS), un aumento del tamaño renal bilateral detectado mediante palpación o en la radiografía abdominal (Figura 4) es altamente sospechoso de poliquistosis (Tabla 3). Mediante el uso de la ecografía, la
identificación de estructuras quísticas nos permite establecer un diagnóstico presuntivo bastante acertado de PKD, y aunque pueden aparecer quistes en los riñones por otras razones esto es extremadamente raro (Figura 5).
■Tratamiento:
Se ha visto que el drenaje de los quistes no enlentece la progresión hacía el fallo renal y hay pocos estudios sobre la eficacia de los IECA en gatos con PKD. No hay evidencias que avalen el uso de IECA en todos los gatos que presentan PKD. La administración de dietas renales sigue los mismos principios que en el manejo de la ERC y deberían iniciarse cuando se alcance el estadio 2.
Con frecuencia los pacientes pueden mostrar episodios de hematuria que remite en reposo y puede ser necesaria la analgesia con el fin de aliviar las molestias. Si existe sospecha de infección quística, lo ideal sería realizar un cultivo del contenido y tratar según los resultados de las pruebas de sensibilidad, teniendo en cuenta que no todos los antibióticos pueden penetrar adecuadamente en el interior de la cavidad quística. En este sentido los antibióticos lipofílicos (quinolonas como marbofloxacino a 2.75-5.5 mg/kg Po una vez al día) suelen ser los preferidos. La duración del tratamiento debería ser de 4-6 semanas. El manejo de posibles infecciones urinarias también es importante, pues pueden ocasionar cuadros sépticos por infección secundaria de los quistes.
- Amiloidosis
La amiloidosis se caracteriza por el depósito extracelular patológico de material proteináceo, formado por la polimerización de subunidades proteicas con una conformación biofísica específica denominada de hoja beta plegada. La amiloidosis más frecuente en los animales domésticos es la reactiva; en la que se produce un depósito tisular de la proteína sérica de fase aguda (amiloide A) en respuesta a la presencia de enfermedades inflamatorias o neoplásicas crónicas. En gatos es poco frecuente y la mayoría de los casos se han descrito en gatos Abisinios, orientales y Siameses, considerándose una predisposición familiar.
En los gatos de raza Abisinia se considera una enfermedad familiar y probablemente heredada de forma autosómica dominante con penetración variable sin predilección sexual. En los pacientes severamente afectados el depósito de amiloide ocurre entre los 9 y 24 meses de edad, fundamentalmente a nivel medular provocando necrosis papilar, fibrosis medular y fallo renal crónico. La ausencia de depósito amiloide a nivel cortical explica la baja frecuencia de proteinuria, y la presentación clínico-patológica más frecuente es la de fallo renal crónico, rápidamente progresivo y renomegalia.
La mayoría de los pacientes muestran sintomatología avanzada a los 3 años de vida. Algunos animales pueden mostrar penetración incompleta y tener una esperanza de vida normal.
En gatos orientales y Siameses con amiloidosis familiar el depósito de amiloide ocurre preferentemente en el hígado y la hemorragia abdominal por rotura de la víscera, suele ser la principal presentación clínica, aunque en algunos individuos puede provocar también ERC. El amiloide identificado en estas razas difiere ligeramente del observado en Abisinios y posiblemente este hecho explique las diferentes localizaciones de los depósitos de amiloide entre razas.
Tras la sospecha clínica y analítica, el diagnóstico solo puede confirmarse mediante la realización de una biopsia renal con la posterior tinción con rojo Congo, observando con microscopio de polarización la birrefringencia verde manzana característica del amiloide. El problema fundamental de la biopsia en estos pacientes, reside en que el amiloide suele depositarse en la médula renal, por lo que en ocasiones las biopsias renales pueden resultar normales porque son biopsias corticales. La amiloidosis es una enfermedad progresiva y los tratamientos propuestos con dimetil sulfóxido y colchicina no parecen dar buenos resultados. El único tratamiento posible una vez identificada la enfermedad, es el manejo de la ERC.
-Otras entidades menos frecuentes
En conclusión, los trastornos renales felinos congénitos, con la excepción de PKD, son poco frecuentes, pero hay que tener en cuenta otras anomalías inusuales para completar este tema.
La displasia renal, se define como un desarrollo desorganizado del parénquima renal debido a una anormal diferenciación, lo que conduce al fallo renal a edades tempranas. Al nacer, en los riñones hay estructuras inmaduras que consisten en tejidos no diferenciados (glomérulos, túbulos fetales y tejido mesenquimatoso y posible tejido cartilaginoso metaplásico) que completan su normal desarrollo antes de los dos meses de vida, sin embargo, en los individuos afectados estos tejidos indiferenciados perdurarán a lo largo de la vida del animal. Las causas de esta anormal nefrogénesis no están plenamente definidas.
Podría ser consecuencia de daños experimentados durante el período fetal o en el período neonatal. Entre las causas potenciales se citan la infección por el virus de la panleucopenia. Se ha descrito un caso aislado en un gato, concretamente en un Bosque de Noruega de 5 meses de edad con poliuria, anorexia y alteraciones laboratoriales indicativas de ERC.
El diagnóstico definitivo se obtiene tan solo mediante estudio histológico con la presencia de al menos tres de los criterios indicados para definir la displasia renal: diferenciación asincrónica del nefrón, presencia de tejido mesenquimatoso, epitelio tubular atípico, conductos metanéfricos persistentes y metaplasia disontogénica.
La extensión de las anormalidades puede afectar a todo el tejido renal o simplemente a una parte, lo que hace que algunos individuos no manifiesten signos clínicos. Aunque macroscópicamente pueden ser normales, en ocasiones los riñones afectados son generalmente más pequeños de lo normal y con estructuras quísticas distribuidas de forma segmentaria o difusa a nivel cortical.
En gatos de raza Abisinia de ambos sexos y familiarmente relacionados se ha descrito una posible nefritis glomerular hereditaria caracterizada por hematuria y proteinuria en grados variables, detectada en todos ellos, entre los 5 y 36 meses. Solo un paciente presentó alteraciones laboratoriales indicativas de fallo renal en el momento de la presentación, y 6 de los 8 animales afectados desarrollaron síndrome nefrótico
con edema periférico.
Histológicamente se detectaron cambios consistentes con glomerulopatía proliferativa focal, no obstante se requieren estudios más profundos que incluyan pruebas inmunohistoquímicas y ultraestructurales para su mejor clasificación e inclusión definitiva dentro de este grupo.
DVM, PhD Maruska Suárez Rey.